30th November 2020 50-year mystery of protein folding solved by AI Protein folding, one of the biggest mysteries in biology, has been solved by artificial intelligence company DeepMind.
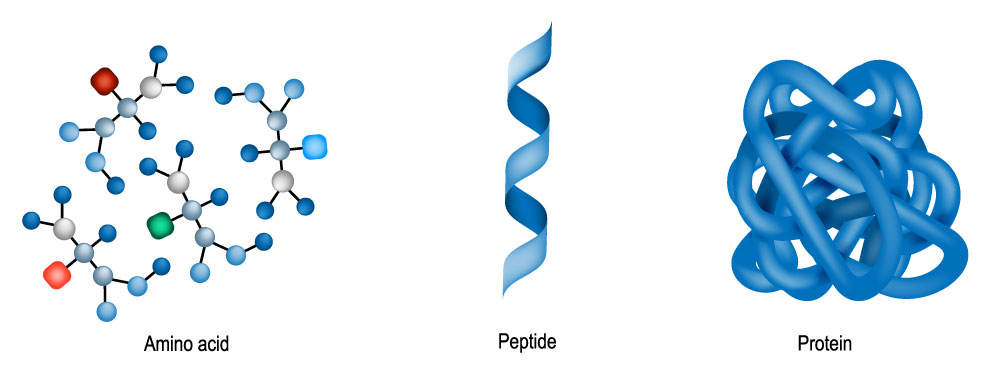
Protein folding is the physical process by which a protein chain acquires its native three-dimensional structure. The process usually occurs on timescales of milliseconds to seconds, although some types can last for minutes or even hours when studied outside a cell. The correct three-dimensional structure is essential for most proteins to function within living organisms. Misfolded proteins can be caused by mutations in the amino acid sequence, or a disruption of the normal folding process by external factors. A number of diseases, along with many allergies, are believed to be the result of misfolded proteins, which makes the study of protein folding of great interest to medical scientists. A protein consists of one or more polypeptides. As illustrated below, these are long peptide chains, themselves containing up to 50 amino acids. In 1969, American molecular biologist, Cyrus Levinthal, noted that because of the very large number of degrees of freedom in an unfolded polypeptide chain, the molecule has an astronomical number of possible spatial arrangements. An estimate as high as 10143 was made in one of his papers.
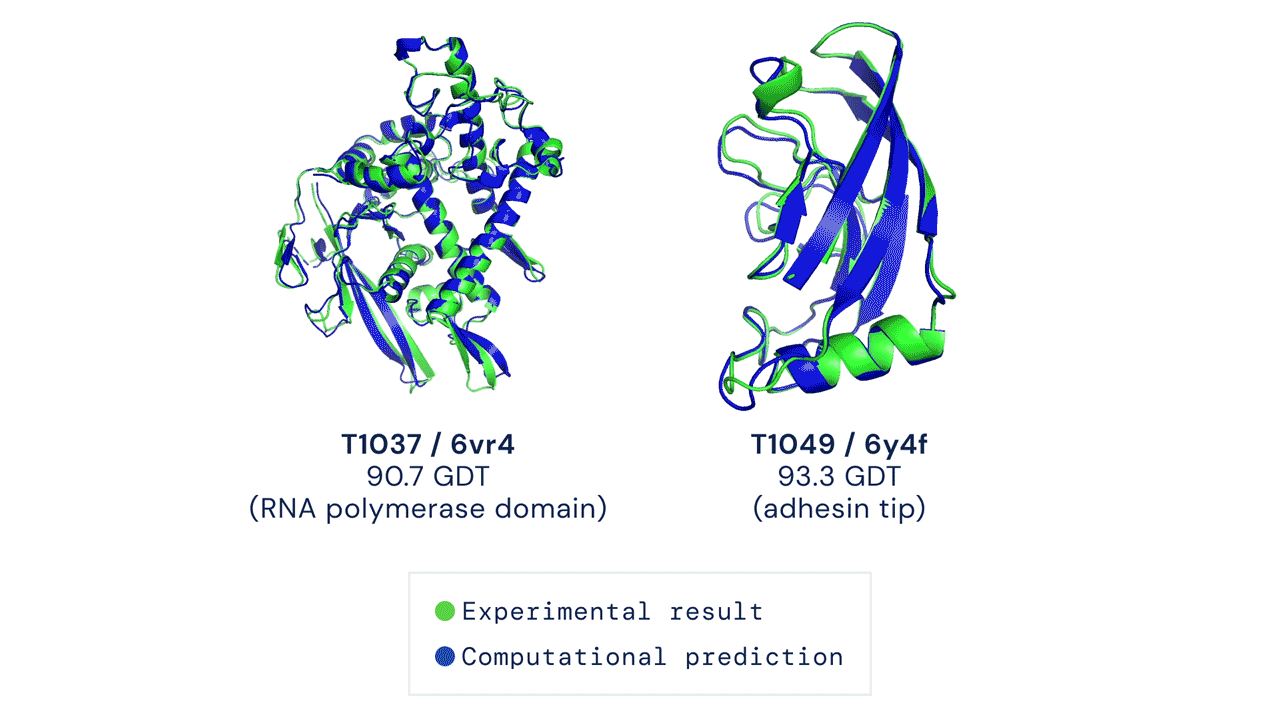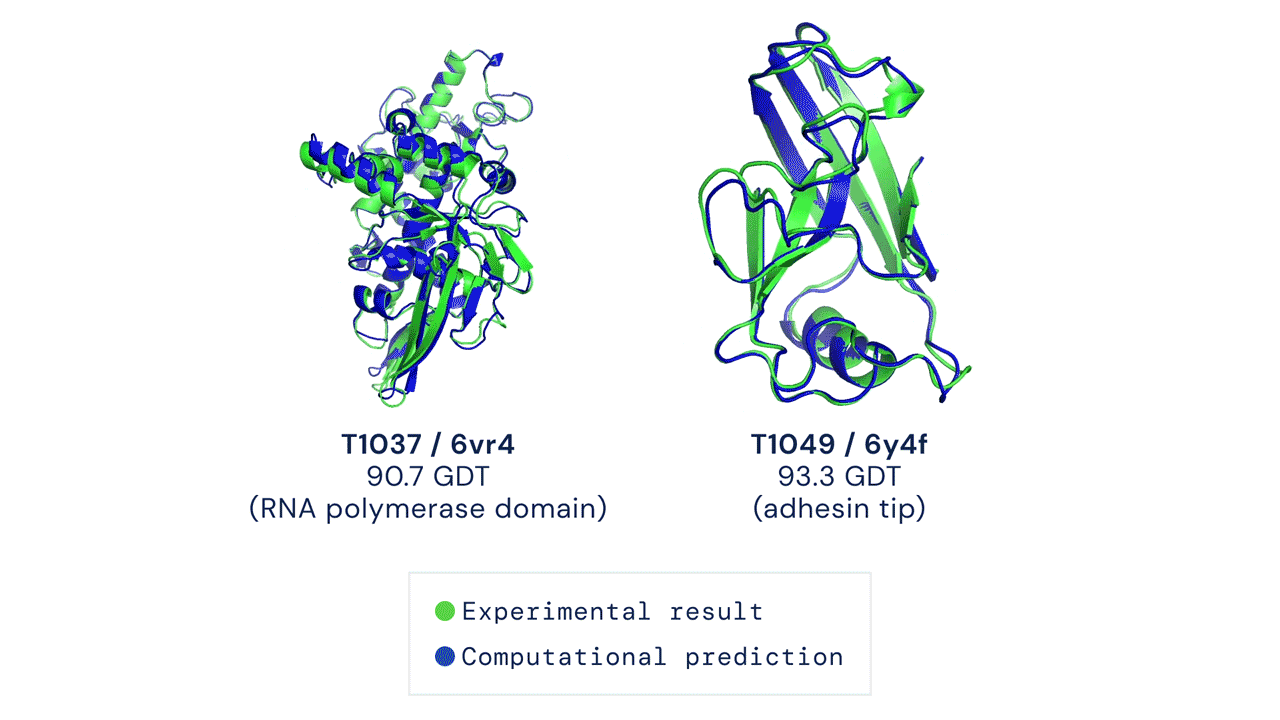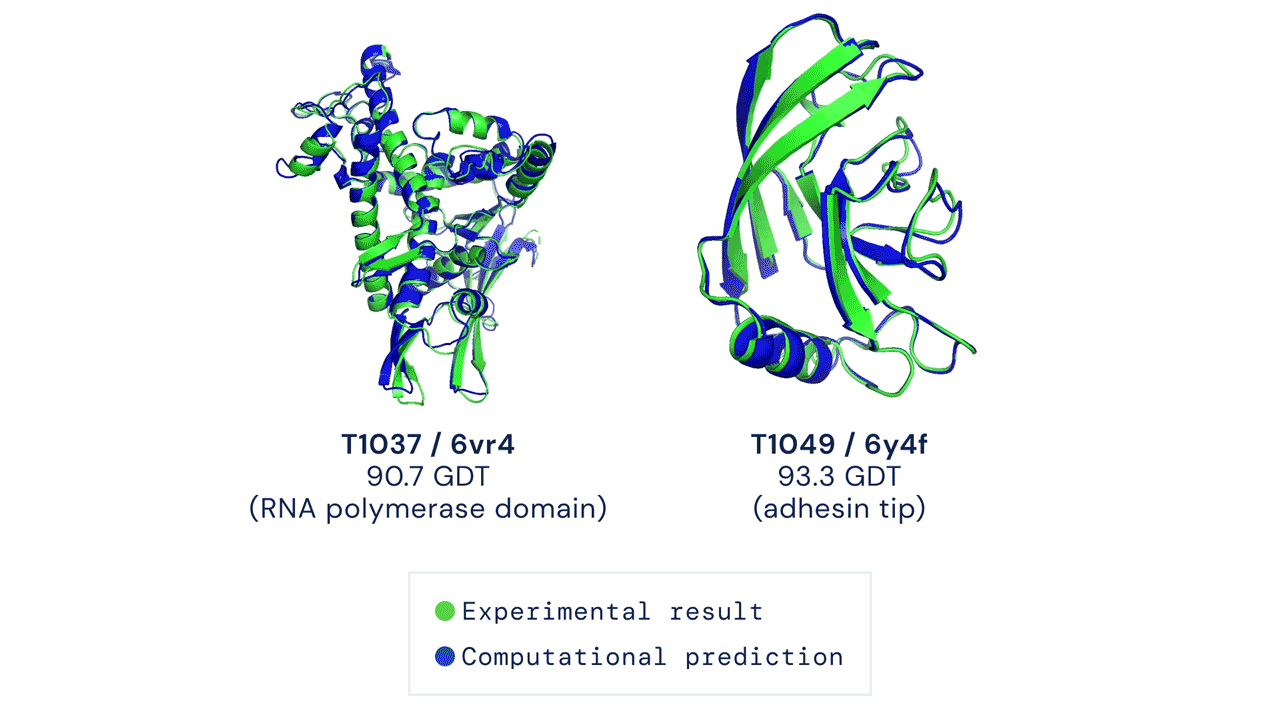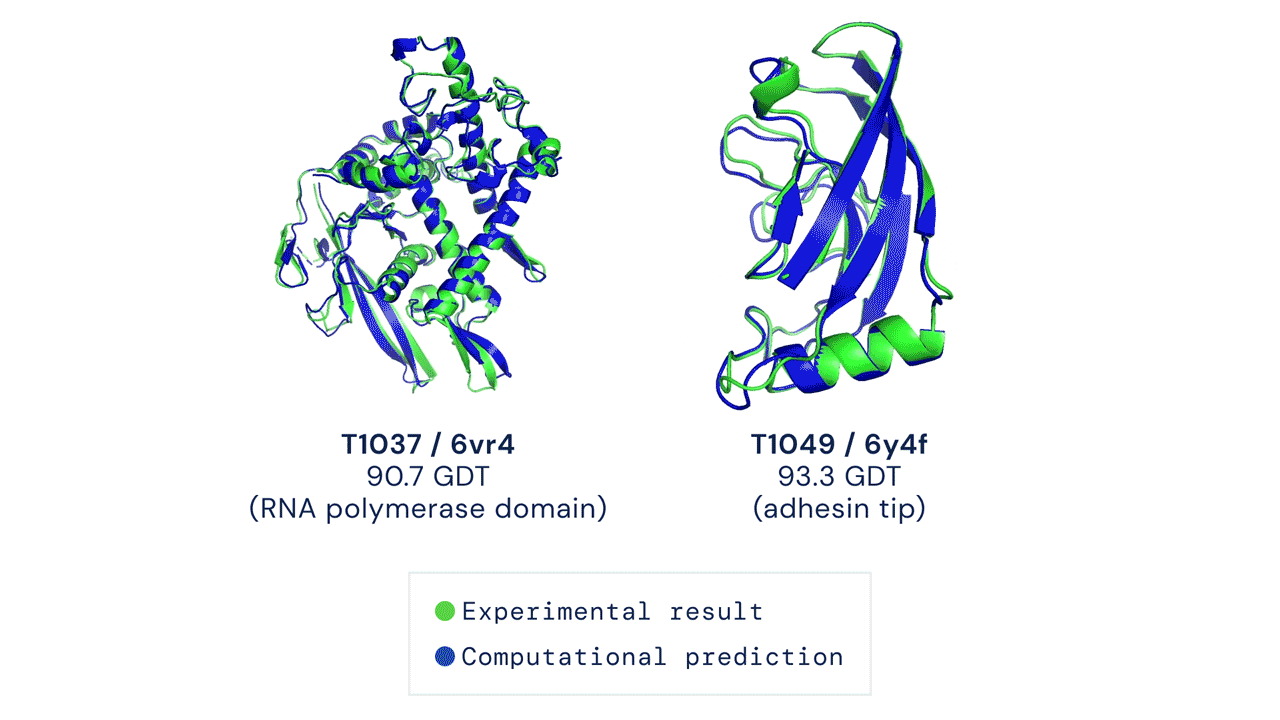
Since then, various experiments have been devised to study protein folding, to reduce its complexity and reveal clues about the process. These methods include nuclear magnetic resonance and X-ray crystallography, as well as newer techniques like cryo-electron microscopy. Protein structure prediction is among the most important goals in bioinformatics and theoretical chemistry; highly important in medicine (for example, in drug design) and biotechnology (for example, in the design of novel enzymes). In a major scientific advance, London-based company DeepMind today announced that the latest version of its AI system, AlphaFold, has produced a solution for the 50-year "protein folding problem". The team at DeepMind used the Critical Assessment of protein Structure Prediction (CASP), an experiment which the global scientific community has tested every two years since 1994. Participants must "blindly" predict the structure of recently discovered proteins, and these predictions are subsequently compared to ground truth experimental data when it becomes available. The main metric for CASP to measure the accuracy of predictions is the Global Distance Test (GDT) which ranges from 0-100. In simple terms, GDT is the percentage of amino acid residues (components of the protein chain) within a threshold distance from the correct position. A score of around 90 GDT is generally considered to be competitive with results obtained from experimental methods. The latest results, published today, show that the AlphaFold AI achieved a median score of 92.4 GDT across all targets. Its average margin of error is comparable to the width of an atom (1.6 Angstroms, or 0.16 nanometres).
"This computational work represents a stunning advance on the protein-folding problem, a 50-year-old grand challenge in biology," said Nobel Laureate and President of the Royal Society, Professor Venki Ramakrishnan. "It has occurred decades before many people in the field would have predicted. It will be exciting to see the many ways in which it will fundamentally change biological research." "AlphaFold is a once-in-a-generation advance, predicting protein structures with incredible speed and precision," said Arthur D. Levinson, PhD, Founder and CEO of Calico. "This leap forward demonstrates how computational methods are poised to transform research in biology and hold much promise for accelerating the drug discovery process." The team at DeepMind trained their AI on 170,000 protein structures from the Protein Data Bank, along with large databases containing protein sequences of unknown structure. For hardware, they used 128 Tensor Processing Units equivalent to between 100 and 200 graphics processing units (GPU) over a few weeks, which the team says is "a relatively modest amount of compute in the context of most large state-of-the-art models used in machine learning." The new system is described as an "attention-based neural network" that attempts to interpret the structure of a "spatial graph", where amino acid residues are the "nodes", and edges connect the residues in close proximity. Through an iterative process, it develops strong predictions of the underlying physical structure of a protein, doing in a matter of days what might take years at a laboratory bench. In addition, it gives estimates on the reliability of different regions of a protein. The unparalleled levels of accuracy by DeepMind enabled them to beat over 100 other teams competing in this year's CASP. "I think it's fair to say this will be very disruptive to the protein-structure-prediction field," said Mohammed AlQuraishi, a computational biologist at Columbia University in New York City and a CASP participant. "I suspect many will leave the field, as the core problem has arguably been solved. It's a breakthrough of the first order, certainly one of the most significant scientific results of my lifetime." The researchers are now working on a paper, to be submitted for publication in a peer-reviewed journal. Additionally, they plan to develop their system in a more scalable way, for wider access to the scientific community. AlphaFold may prove useful in future pandemic response efforts, for example, by enabling treatments to arrive sooner. "As well as accelerating understanding of known diseases, we're excited about the potential for these techniques to explore the hundreds of millions of proteins we don't currently have models for – a vast terrain of unknown biology," says the team. "Among the undetermined proteins may be some with new and exciting functions and – just as a telescope helps us see deeper into the unknown universe – techniques like AlphaFold may help us find them." --- Follow us: Twitter | Facebook | Instagram | YouTube
Comments »
If you enjoyed this article, please consider sharing it:
|